Spatiotemporal Regulatory Analysis of Tumors#
Data preprocessing#
The spatial transcriptomics data required for the analysis, specifically from OSCC patients 2, 3, and 5, can be downloaded from http://www.pboselab.ca/spatial_OSCC/. Additional files are available at https://figshare.com/articles/dataset/Spatial_transcriptomics_reveals_distinct_and_conserved_tumor_core_and_edge_architectures_that_predict_survival_and_targeted_therapy_response_/20304456/1. We implemented a function for data preprocessing to integrate information from datasets of different formats, including RNA velocity analysis.
[1]:
import pandas as pd
import scvelo as scv
import scanpy as sc
import numpy as np
scv.logging.print_version()
import matplotlib.pyplot as plt
import seaborn as sb
import matplotlib as mpl
import seaborn as sns
E:\Anaconda\envs\sparcl\lib\site-packages\tqdm\auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Running scvelo 0.2.5 (python 3.8.13) on 2025-06-09 16:47.
[2]:
scv.settings.verbosity = 3
scv.settings.presenter_view = True
scv.set_figure_params('scvelo')
[3]:
def Process(file_wd,sample):
sc_adata = scv.read(file_wd+sample+'/'+sample+'_possorted_genome_bam.loom', cache=False)
sp_adata = sc.read_visium(file_wd+sample+'/')
anno = pd.read_csv(file_wd+sample+'/'+'annotation.csv',index_col=0)
match=[]
for bar in sc_adata.obs_names:
match.append(bar.split(':')[1])
sc_index=[]
for bar in match:
sc_index.append(bar.split('x')[0])
sp_index=[]
for bar in sp_adata.obs_names:
sp_index.append(bar.split('-')[0])
sc_adata.obs_names=sc_index
sp_adata.obs_names=sp_index
anno_index=[]
for bar in anno.index:
anno_index.append(bar.split('-')[0])
anno.index = anno_index
sp_adata.obs['celltype']=anno['pathologist_anno.x']
sp_adata.obs['cluster']=anno['cluster_annotations']
all_sp_adata=sp_adata
sp_adata=sp_adata[sc_adata.obs_names,:]
common=list(set(anno_index)&set(sc_index))
anno=anno.loc[common]
sc_adata.obsm['spatial']=sp_adata.obsm['spatial'].astype('float32')
sc_adata.obs['cluster_annotations']=anno['cluster_annotations']
sc_adata=sc_adata[sc_adata.obs['cluster_annotations'].isin(['core','edge','transitory']),:]
scv.pp.filter_and_normalize(sc_adata)
scv.pp.moments(sc_adata)
scv.tl.recover_dynamics(sc_adata,n_jobs = 10)
scv.tl.velocity_graph(sc_adata)
scv.tl.velocity(sc_adata, mode='dynamical')
scv.tl.velocity_graph(sc_adata)
scv.tl.latent_time(sc_adata)
scv.tl.velocity_confidence(sc_adata)
sc_adata.obs['celltype']=anno['pathologist_anno.x']
return list((sc_adata,all_sp_adata))
[4]:
all_data=dict()
samples=['s2','s3','s5']
data_file='E:/Oral_squamous_cell_carcinoma/'
for sample in samples:
all_data[sample]=Process(data_file,sample)
all_data[sample][1].write_h5ad(data_file+sample+'/'+'sp_adata.h5ad')
all_data[sample][0].write_h5ad(data_file+sample+'/'+'sc_adata.h5ad')
[5]:
samples=['s2','s3','s5']
data_file='E:/Oral_squamous_cell_carcinoma/'
adata_k={}
for sample in samples:
adata_k[sample] = sc.read_h5ad(data_file+sample+'/sp_adata.h5ad')
adata_k[sample]
adata_k[sample].obs['cancer']='YES'
index=adata_k[sample][~adata_k[sample].obs['cluster'].isin(['core','edge','transitory'])].obs_names
adata_k[sample].obs.loc[index,'cancer']='NO'
fig, axs = plt.subplots(1,3,sharex=True, sharey=True,figsize=(16, 4))
sc.pl.spatial(
adata_k['s2'],
basis= 'spatial',
frameon=False,
color='cancer',
size=1,
colorbar_loc=None,
show=False,
title='s2',
ax=axs[0]
)
sc.pl.spatial(
adata_k['s3'],
basis= 'spatial',
frameon=False,
color='cancer',
size=1,
colorbar_loc=None,
show=False,
title='s3',
ax=axs[1]
)
sc.pl.spatial(
adata_k['s5'],
basis= 'spatial',
frameon=False,
color='cancer',
size=1,
colorbar_loc=None,
show=False,
title='s5',
ax=axs[2]
)
[5]:
[<AxesSubplot: title={'center': 's5'}, xlabel='spatial1', ylabel='spatial2'>]

Batch correction#
We performed batch correction using patient identity as the batch label.
[6]:
import HieDiff as hd
import anndata as ad
[7]:
s2 = sc.read_h5ad('E:/Oral_squamous_cell_carcinoma/s2/sc_adata.h5ad')
s5 = sc.read_h5ad('E:/Oral_squamous_cell_carcinoma/s5/sc_adata.h5ad')
s3 = sc.read_h5ad('E:/Oral_squamous_cell_carcinoma/s3/sc_adata.h5ad')
[8]:
palette1={'core':'#98df8a',
'edge':'#ff9896',
'transitory':'#c5b0d5'}
[9]:
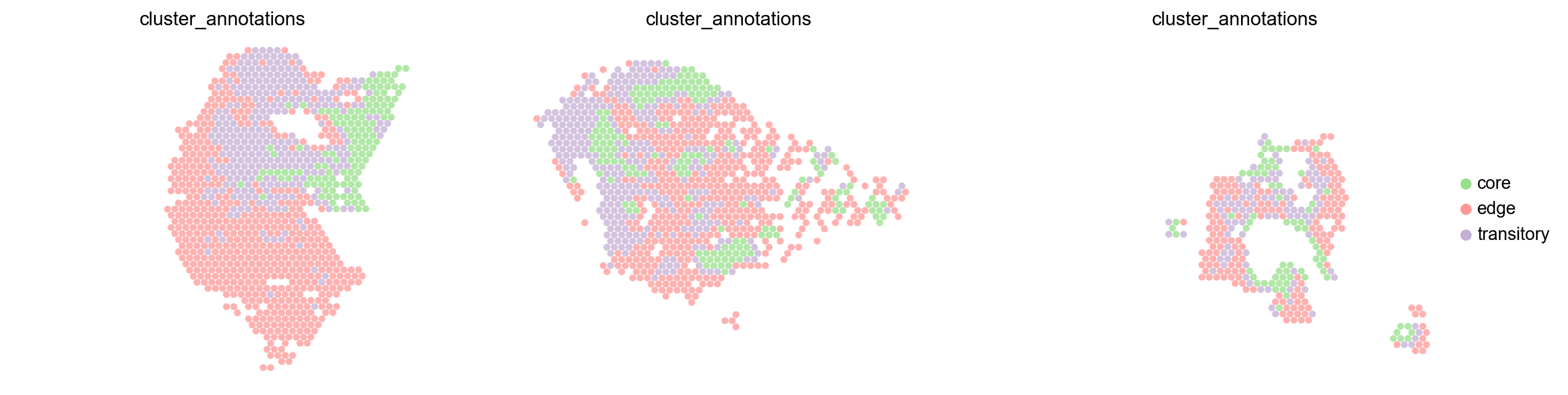
fig, axs = plt.subplots(1,3,sharex=True, sharey=True,figsize=(16, 4))
sc.pl.embedding(
s2,
basis= 'spatial',
frameon=False,
color='cluster_annotations',
size=75,
palette=palette1,
colorbar_loc=None,
show=False,
ax=axs[0],
legend_loc=None,
alpha=0.75
)
sc.pl.embedding(
s5,
basis= 'spatial',
frameon=False,
color='cluster_annotations',
size=75,
palette=palette1,
colorbar_loc=None,
show=False,
ax=axs[1],
legend_loc=None,
alpha=0.75
)
sc.pl.embedding(
s3,
basis= 'spatial',
frameon=False,
color='cluster_annotations',
size=75,
palette=palette1,
colorbar_loc=None,
show=False,
ax=axs[2],
alpha=0.75
)
[9]:
<AxesSubplot: title={'center': 'cluster_annotations'}, xlabel='spatial1', ylabel='spatial2'>
[10]:
s2.obs['sample']='s2'
s5.obs['sample']='s5'
s3.obs['sample']='s3'
s2.obs_names='sample2'+s2.obs_names
s5.obs_names='sample5'+s5.obs_names
s3.obs_names='sample3'+s3.obs_names
adata=ad.concat([s2,s5,s3],uns_merge='first',merge='first')
sc.pp.filter_genes(adata,min_counts=15)
sc.pp.filter_genes(adata,min_cells=15)
[11]:
sc.pp.highly_variable_genes(adata, n_top_genes=6000, flavor='seurat_v3',batch_key='sample')
[12]:
adata1 = adata[:, adata.var['highly_variable']]
[13]:
#adata1 = sc.read_h5ad('E:/Oral_squamous_cell_carcinoma/adata.h5ad')
sc.pp.pca(adata1)
sc.pp.neighbors(adata1)
sc.tl.umap(adata1,min_dist=1)
fig, axs = plt.subplots(1,2,figsize=(4, 1.75))
sc.pl.embedding(
adata1,
basis= 'umap',
color='cluster_annotations',
size=10,
palette=palette1,
colorbar_loc=None,
show=False,
ax=axs[0],
alpha=0.75
)
sc.pl.embedding(
adata1,
basis= 'umap',
color='sample',
size=10,
colorbar_loc=None,
show=False,
ax=axs[1],
alpha=1
)
[13]:
<AxesSubplot: title={'center': 'sample'}, xlabel='UMAP1', ylabel='UMAP2'>

[14]:
hd.run_HieDiff(adata1, n_epochs=5000, n_hidden=256, n_latent=128, batch_key='sample')
[15]:
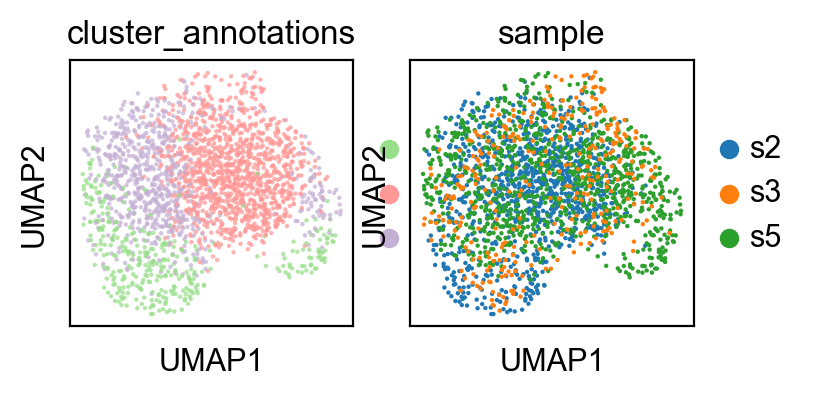
sc.pp.neighbors(adata1,use_rep='qz')
sc.tl.umap(adata1,min_dist=1)
fig, axs = plt.subplots(1,2,figsize=(4, 1.75))
sc.pl.embedding(
adata1,
basis= 'umap',
color='cluster_annotations',
size=10,
palette=palette1,
colorbar_loc=None,
show=False,
ax=axs[0],
alpha=0.75
)
sc.pl.embedding(
adata1,
basis= 'umap',
color='sample',
size=10,
colorbar_loc=None,
show=False,
ax=axs[1],
alpha=1
)
[15]:
<AxesSubplot: title={'center': 'sample'}, xlabel='UMAP1', ylabel='UMAP2'>
[16]:
adata1.X=adata1.layers['x4']
Regulatory network inference#
Based on the inferred gene co-expression matrix W, we performed regulon inference and quantified the activity of each regulon across spatial locations.
[17]:
from yaml import Loader, Dumper
import glob
from typing import Optional, Sequence
from anndata import AnnData
from yaml import load, dump
try:
from yaml import CLoader as Loader, CDumper as Dumper
except ImportError:
from yaml import Loader, Dumper
from pyscenic.rnkdb import FeatherRankingDatabase as RankingDatabase
from pyscenic.utils import modules_from_adjacencies
from pyscenic.prune import prune2df, df2regulons
from pyscenic.aucell import aucell as pyscenic_aucell
[18]:
MOTIF_ANNOTATIONS_FNAME='E:/CisTarget/motifs-v9-nr.hgnc-m0.001-o0.0.tbl'
tf_names=np.array((pd.read_table('E:/CisTarget/hs_hgnc_tfs.txt',header=None).iloc[:,0]))
DATABASES_GLOB='E:/CisTarget/hg19-*.mc9nr.feather'
db_fnames = glob.glob(DATABASES_GLOB)
[19]:
hd.regulons(adata1, tf_names, MOTIF_ANNOTATIONS_FNAME, db_fnames, neighbors_var_key='W')
[20]:
hd.aucell(adata1, normalize=True)
[21]:
adata1.obsm['aucell'] /= np.quantile(adata1.obsm['aucell'], q=0.999, axis=0, keepdims=True)
adata1.obsm['aucell'][adata1.obsm['aucell'] > 1] = 1
[22]:
adata1
[22]:
AnnData object with n_obs × n_vars = 2267 × 6000
obs: 'cluster_annotations', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts', 'velocity_self_transition', 'root_cells', 'end_points', 'velocity_pseudotime', 'latent_time', 'velocity_length', 'velocity_confidence', 'velocity_confidence_transition', 'celltype', 'sample'
var: 'Accession', 'Chromosome', 'End', 'Start', 'Strand', 'gene_count_corr', 'fit_r2', 'fit_alpha', 'fit_beta', 'fit_gamma', 'fit_t_', 'fit_scaling', 'fit_std_u', 'fit_std_s', 'fit_likelihood', 'fit_u0', 'fit_s0', 'fit_pval_steady', 'fit_steady_u', 'fit_steady_s', 'fit_variance', 'fit_alignment_scaling', 'velocity_gamma', 'velocity_qreg_ratio', 'velocity_r2', 'velocity_genes', 'n_cells', 'n_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'VAE', 'W', 'aucell', 'cluster_annotations_colors', 'hvg', 'log1p', 'neighbors', 'pca', 'recover_dynamics', 'regulons', 'sample_colors', 'spatial_reconstruction', 'umap', 'velocity_graph', 'velocity_graph_neg', 'velocity_params'
obsm: 'X_pca', 'X_umap', 'aucell', 'qz', 'spatial'
varm: 'PCs', 'loss'
layers: 'Ms', 'Mu', 'ambiguous', 'counts', 'fit_t', 'fit_tau', 'fit_tau_', 'log1p', 'matrix', 'spliced', 'unspliced', 'variance_velocity', 'velocity', 'velocity_u', 'x4'
obsp: 'connectivities', 'distances'
varp: 'W'
[23]:
def adata_split(adata, batch_key_list, batch_key="batch"):
adata_list = {}
for i in batch_key_list:
print(i)
index=np.flatnonzero(adata.obs[batch_key]==str(i))
adata_list[str(i)]=adata[index,]
return adata_list
[24]:
adata=adata_split(adata1,['s2','s5','s3'],'sample')
s2
s5
s3
[25]:
adata
[25]:
{'s2': View of AnnData object with n_obs × n_vars = 983 × 6000
obs: 'cluster_annotations', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts', 'velocity_self_transition', 'root_cells', 'end_points', 'velocity_pseudotime', 'latent_time', 'velocity_length', 'velocity_confidence', 'velocity_confidence_transition', 'celltype', 'sample'
var: 'Accession', 'Chromosome', 'End', 'Start', 'Strand', 'gene_count_corr', 'fit_r2', 'fit_alpha', 'fit_beta', 'fit_gamma', 'fit_t_', 'fit_scaling', 'fit_std_u', 'fit_std_s', 'fit_likelihood', 'fit_u0', 'fit_s0', 'fit_pval_steady', 'fit_steady_u', 'fit_steady_s', 'fit_variance', 'fit_alignment_scaling', 'velocity_gamma', 'velocity_qreg_ratio', 'velocity_r2', 'velocity_genes', 'n_cells', 'n_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'VAE', 'W', 'aucell', 'cluster_annotations_colors', 'hvg', 'log1p', 'neighbors', 'pca', 'recover_dynamics', 'regulons', 'sample_colors', 'spatial_reconstruction', 'umap', 'velocity_graph', 'velocity_graph_neg', 'velocity_params'
obsm: 'X_pca', 'X_umap', 'aucell', 'qz', 'spatial'
varm: 'PCs', 'loss'
layers: 'Ms', 'Mu', 'ambiguous', 'counts', 'fit_t', 'fit_tau', 'fit_tau_', 'log1p', 'matrix', 'spliced', 'unspliced', 'variance_velocity', 'velocity', 'velocity_u', 'x4'
obsp: 'connectivities', 'distances'
varp: 'W',
's5': View of AnnData object with n_obs × n_vars = 942 × 6000
obs: 'cluster_annotations', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts', 'velocity_self_transition', 'root_cells', 'end_points', 'velocity_pseudotime', 'latent_time', 'velocity_length', 'velocity_confidence', 'velocity_confidence_transition', 'celltype', 'sample'
var: 'Accession', 'Chromosome', 'End', 'Start', 'Strand', 'gene_count_corr', 'fit_r2', 'fit_alpha', 'fit_beta', 'fit_gamma', 'fit_t_', 'fit_scaling', 'fit_std_u', 'fit_std_s', 'fit_likelihood', 'fit_u0', 'fit_s0', 'fit_pval_steady', 'fit_steady_u', 'fit_steady_s', 'fit_variance', 'fit_alignment_scaling', 'velocity_gamma', 'velocity_qreg_ratio', 'velocity_r2', 'velocity_genes', 'n_cells', 'n_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'VAE', 'W', 'aucell', 'cluster_annotations_colors', 'hvg', 'log1p', 'neighbors', 'pca', 'recover_dynamics', 'regulons', 'sample_colors', 'spatial_reconstruction', 'umap', 'velocity_graph', 'velocity_graph_neg', 'velocity_params'
obsm: 'X_pca', 'X_umap', 'aucell', 'qz', 'spatial'
varm: 'PCs', 'loss'
layers: 'Ms', 'Mu', 'ambiguous', 'counts', 'fit_t', 'fit_tau', 'fit_tau_', 'log1p', 'matrix', 'spliced', 'unspliced', 'variance_velocity', 'velocity', 'velocity_u', 'x4'
obsp: 'connectivities', 'distances'
varp: 'W',
's3': View of AnnData object with n_obs × n_vars = 342 × 6000
obs: 'cluster_annotations', 'initial_size_unspliced', 'initial_size_spliced', 'initial_size', 'n_counts', 'velocity_self_transition', 'root_cells', 'end_points', 'velocity_pseudotime', 'latent_time', 'velocity_length', 'velocity_confidence', 'velocity_confidence_transition', 'celltype', 'sample'
var: 'Accession', 'Chromosome', 'End', 'Start', 'Strand', 'gene_count_corr', 'fit_r2', 'fit_alpha', 'fit_beta', 'fit_gamma', 'fit_t_', 'fit_scaling', 'fit_std_u', 'fit_std_s', 'fit_likelihood', 'fit_u0', 'fit_s0', 'fit_pval_steady', 'fit_steady_u', 'fit_steady_s', 'fit_variance', 'fit_alignment_scaling', 'velocity_gamma', 'velocity_qreg_ratio', 'velocity_r2', 'velocity_genes', 'n_cells', 'n_counts', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'VAE', 'W', 'aucell', 'cluster_annotations_colors', 'hvg', 'log1p', 'neighbors', 'pca', 'recover_dynamics', 'regulons', 'sample_colors', 'spatial_reconstruction', 'umap', 'velocity_graph', 'velocity_graph_neg', 'velocity_params'
obsm: 'X_pca', 'X_umap', 'aucell', 'qz', 'spatial'
varm: 'PCs', 'loss'
layers: 'Ms', 'Mu', 'ambiguous', 'counts', 'fit_t', 'fit_tau', 'fit_tau_', 'log1p', 'matrix', 'spliced', 'unspliced', 'variance_velocity', 'velocity', 'velocity_u', 'x4'
obsp: 'connectivities', 'distances'
varp: 'W'}
[26]:
s2_r=adata['s2'].obsm['aucell'].loc[:,~(adata['s2'].obsm['aucell']==0).all(axis=0)]
s5_r=adata['s5'].obsm['aucell'].loc[:,~(adata['s5'].obsm['aucell']==0).all(axis=0)]
s3_r=adata['s3'].obsm['aucell'].loc[:,~(adata['s3'].obsm['aucell']==0).all(axis=0)]
[27]:
s2_r=s2_r.dropna(axis=1,how='any')
s5_r=s5_r.dropna(axis=1,how='any')
s3_r=s3_r.dropna(axis=1,how='any')
[28]:
s2_regulon=sc.AnnData(s2_r)
s5_regulon=sc.AnnData(s5_r)
s3_regulon=sc.AnnData(s3_r)
[29]:
palette1=['#98df8a','#ff9896','#c5b0d5']
s2_regulon.uns['cluster_annotations_colors']=palette1
s5_regulon.uns['cluster_annotations_colors']=palette1
s3_regulon.uns['cluster_annotations_colors']=palette1
[30]:
s2_regulon.obs=s2.obs
s5_regulon.obs=s5.obs
s3_regulon.obs=s3.obs
s2_regulon.obsm=s2.obsm
s5_regulon.obsm=s5.obsm
s3_regulon.obsm=s3.obsm
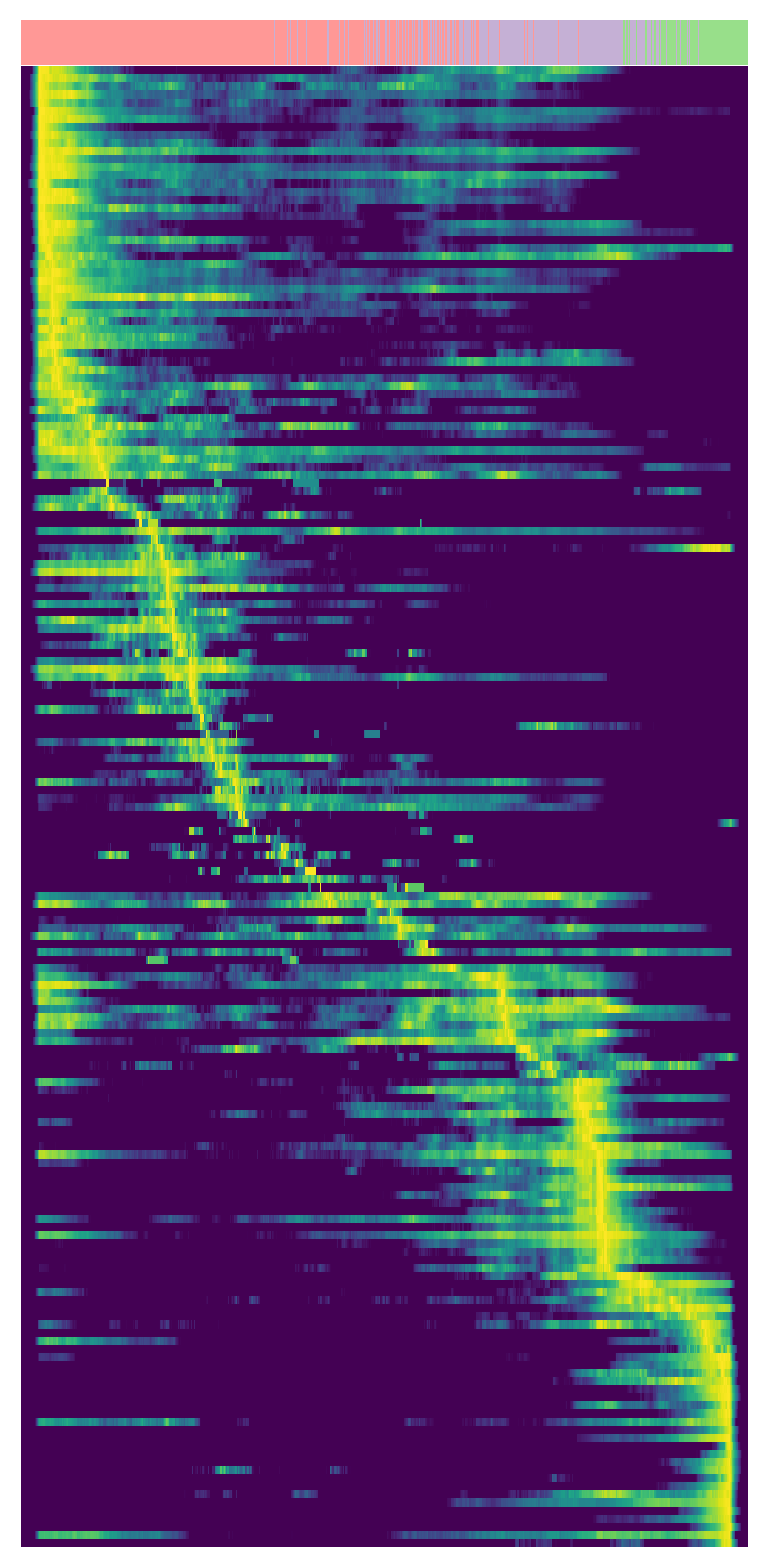
Spatiotemporal dynamics of regulons#
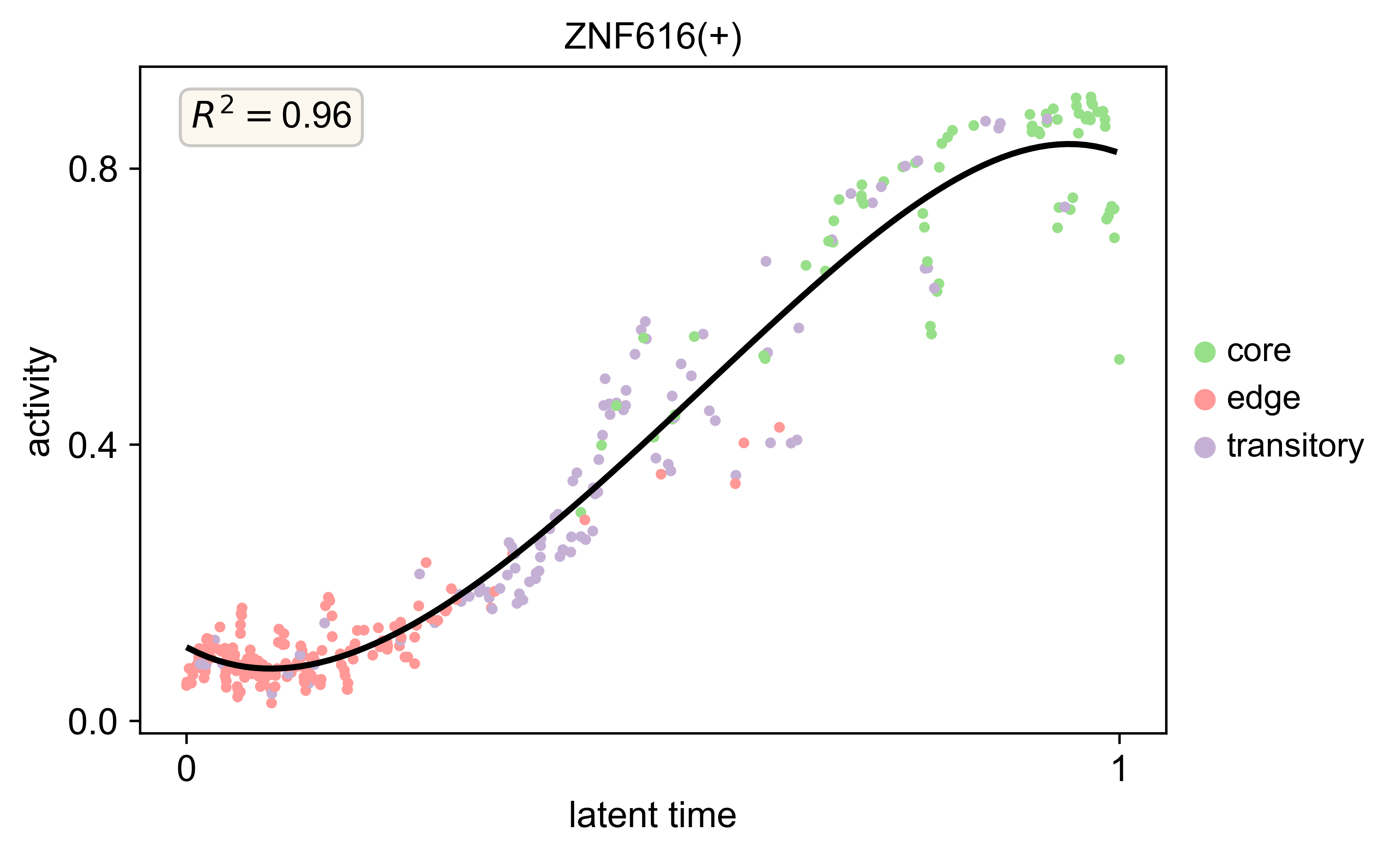
We ordered the activity of each spot along the pseudotime inferred from RNA velocity analysis, generating an activity–pseudotime heatmap to identify regulons that may play critical roles in tumor state transitions.
[31]:
import numpy as np
import pandas as pd
from scipy.sparse import issparse
from scvelo import logging as logg
from scvelo.plotting.utils import (
interpret_colorkey,
is_categorical,
savefig_or_show,
set_colors_for_categorical_obs,
strings_to_categoricals,
to_list,
)
def heatmap(
adata,
var_names,
sortby="latent_time",
layer="Ms",
color_map="viridis",
col_color=None,
palette="viridis",
n_convolve=30,
standard_scale=0,
sort=True,
colorbar=None,
col_cluster=False,
row_cluster=False,
context=None,
font_scale=None,
figsize=(8, 4),
show=None,
save=None,
**kwargs,
):
"""Plot time series for genes as heatmap.
Arguments:
---------
adata: :class:`~anndata.AnnData`
Annotated data matrix.
var_names: `str`, list of `str`
Names of variables to use for the plot.
sortby: `str` (default: `'latent_time'`)
Observation key to extract time data from.
layer: `str` (default: `'Ms'`)
Layer key to extract count data from.
color_map: `str` (default: `'viridis'`)
String denoting matplotlib color map.
col_color: `str` or list of `str` (default: `None`)
String denoting matplotlib color map to use along the columns.
palette: list of `str` (default: `'viridis'`)
Colors to use for plotting groups (categorical annotation).
n_convolve: `int` or `None` (default: `30`)
If `int` is given, data is smoothed by convolution
along the x-axis with kernel size n_convolve.
standard_scale : `int` or `None` (default: `0`)
Either 0 (rows) or 1 (columns). Whether or not to standardize that dimension
(each row or column), subtract minimum and divide each by its maximum.
sort: `bool` (default: `True`)
Wether to sort the expression values given by xkey.
colorbar: `bool` or `None` (default: `None`)
Whether to show colorbar.
{row,col}_cluster : `bool` or `None`
If True, cluster the {rows, columns}.
context : `None`, or one of {paper, notebook, talk, poster}
A dictionary of parameters or the name of a preconfigured set.
font_scale : float, optional
Scaling factor to scale the size of the font elements.
figsize: tuple (default: `(8,4)`)
Figure size.
show: `bool`, optional (default: `None`)
Show the plot, do not return axis.
save: `bool` or `str`, optional (default: `None`)
If `True` or a `str`, save the figure. A string is appended to the default
filename. Infer the filetype if ending on {'.pdf', '.png', '.svg'}.
kwargs:
Arguments: passed to seaborns clustermap,
e.g., set `yticklabels=True` to display all gene names in all rows.
Returns
-------
If `show==False` a `matplotlib.Axis`
"""
import seaborn as sns
var_names = [name for name in var_names if name in adata.var_names]
tkey, xkey = kwargs.pop("tkey", sortby), kwargs.pop("xkey", layer)
time = adata.obs[tkey].values
time = time[np.isfinite(time)]
X = (
adata[:, var_names].layers[xkey]
if xkey in adata.layers.keys()
else adata[:, var_names].X
)
if issparse(X):
X = X.A
df = pd.DataFrame(X[np.argsort(time)], columns=var_names)
if n_convolve is not None:
weights = np.ones(n_convolve) / n_convolve
for gene in var_names:
# TODO: Handle exception properly
try:
df[gene] = np.convolve(df[gene].values, weights, mode="same")
except ValueError as e:
logg.info(f"Skipping variable {gene}: {e}")
pass # e.g. all-zero counts or nans cannot be convolved
if sort:
max_sort = np.argsort(np.argmax(df.values, axis=0))
df = pd.DataFrame(df.values[:, max_sort], columns=df.columns[max_sort])
strings_to_categoricals(adata)
if col_color is not None:
col_colors = to_list(col_color)
col_color = []
for _, col in enumerate(col_colors):
if not is_categorical(adata, col):
obs_col = adata.obs[col]
cat_col = np.round(obs_col / np.max(obs_col), 2) * np.max(obs_col)
adata.obs[f"{col}_categorical"] = pd.Categorical(cat_col)
col += "_categorical"
#set_colors_for_categorical_obs(adata, col, palette)
col_color.append(interpret_colorkey(adata, col)[np.argsort(time)])
if "dendrogram_ratio" not in kwargs:
kwargs["dendrogram_ratio"] = (
0.1 if row_cluster else 0,
0.2 if col_cluster else 0,
)
if "cbar_pos" not in kwargs or not colorbar:
kwargs["cbar_pos"] = None
kwargs.update(
{
"col_colors": col_color,
"col_cluster": col_cluster,
"row_cluster": row_cluster,
"cmap": color_map,
"xticklabels": False,
"standard_scale": standard_scale,
"figsize": figsize,
}
)
args = {}
if font_scale is not None:
args = {"font_scale": font_scale}
context = context or "notebook"
with sns.plotting_context(context=context, **args):
# TODO: Remove exception by requiring appropriate seaborn version
try:
cm = sns.clustermap(df.T, **kwargs)
except ImportWarning:
logg.warn("Please upgrade seaborn with `pip install -U seaborn`.")
kwargs.pop("dendrogram_ratio")
kwargs.pop("cbar_pos")
cm = sns.clustermap(df.T, **kwargs)
savefig_or_show("heatmap", save=save, show=show)
if show is False:
return cm
[32]:
gene=list(s2_regulon.var_names&s5_regulon.var_names&s3_regulon.var_names)
[33]:
scv.pl.heatmap(s2_regulon, var_names=gene,n_convolve=50,yticklabels=False,
sortby='latent_time',vmin=0.70,vmax=1.0,figsize=(4,8),col_color='cluster_annotations')
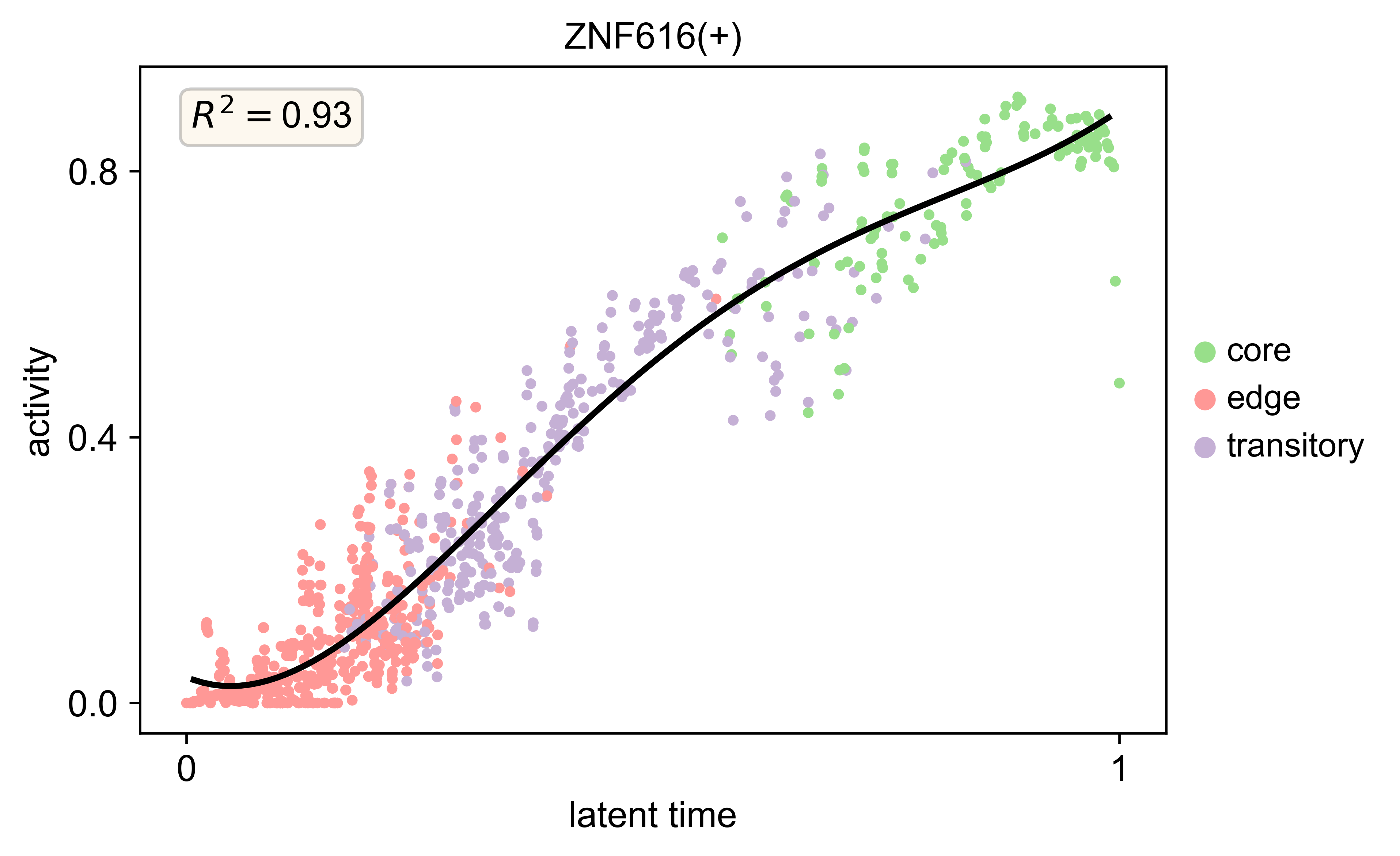
[34]:
scv.pl.scatter(s2_regulon, x='latent_time', y=['ZNF616(+)'],color='cluster_annotations',add_polyfit=5,n_convolve=5,linewidth=2,
legend_loc='right margin',ylabel='activity',dpi=300,size=50,show=True)
scv.pl.scatter(s5_regulon, x='latent_time', y=['ZNF616(+)'],color='cluster_annotations',add_polyfit=5,n_convolve=5,linewidth=2,
legend_loc='right margin',ylabel='activity',dpi=300,size=50,show=True)
scv.pl.scatter(s3_regulon, x='latent_time', y=['ZNF616(+)'],color='cluster_annotations',add_polyfit=5,n_convolve=5,linewidth=2,
legend_loc='right margin',ylabel='activity',dpi=300,size=50,show=True)

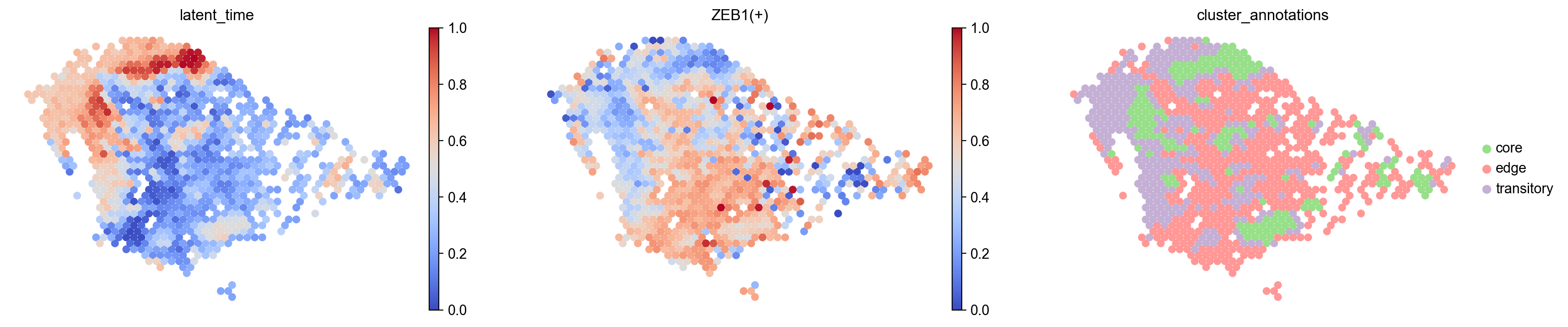
[35]:
sc.pl.embedding(
s5_regulon,
basis= 'spatial',
frameon=False,
color=['latent_time','ZEB1(+)','cluster_annotations'],
size=150,
cmap='coolwarm',
show=False
)
[35]:
[<AxesSubplot: title={'center': 'latent_time'}, xlabel='spatial1', ylabel='spatial2'>,
<AxesSubplot: title={'center': 'ZEB1(+)'}, xlabel='spatial1', ylabel='spatial2'>,
<AxesSubplot: title={'center': 'cluster_annotations'}, xlabel='spatial1', ylabel='spatial2'>]
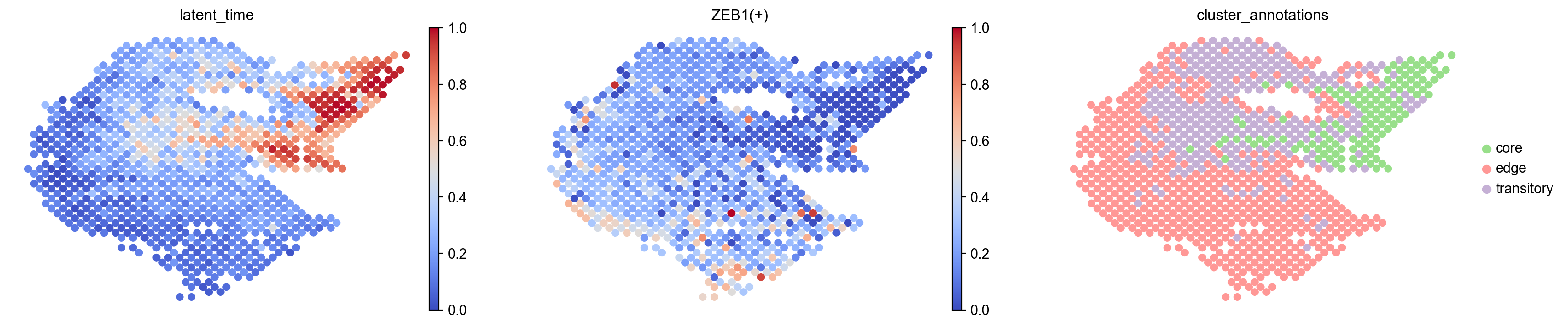
[36]:
sc.pl.embedding(
s3_regulon,
basis= 'spatial',
frameon=False,
color=['latent_time','ZEB1(+)','cluster_annotations'],
size=150,
cmap='coolwarm',
show=False
)
[36]:
[<AxesSubplot: title={'center': 'latent_time'}, xlabel='spatial1', ylabel='spatial2'>,
<AxesSubplot: title={'center': 'ZEB1(+)'}, xlabel='spatial1', ylabel='spatial2'>,
<AxesSubplot: title={'center': 'cluster_annotations'}, xlabel='spatial1', ylabel='spatial2'>]

[37]:
sc.pl.embedding(
s2_regulon,
basis= 'spatial',
frameon=False,
color=['latent_time','ZEB1(+)','cluster_annotations'],
size=150,
cmap='coolwarm',
show=False
)
[37]:
[<AxesSubplot: title={'center': 'latent_time'}, xlabel='spatial1', ylabel='spatial2'>,
<AxesSubplot: title={'center': 'ZEB1(+)'}, xlabel='spatial1', ylabel='spatial2'>,
<AxesSubplot: title={'center': 'cluster_annotations'}, xlabel='spatial1', ylabel='spatial2'>]
[ ]: